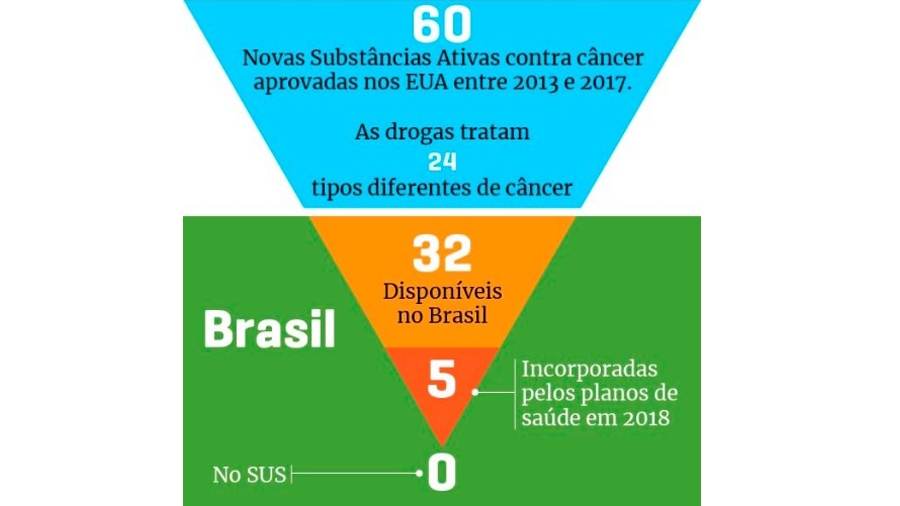
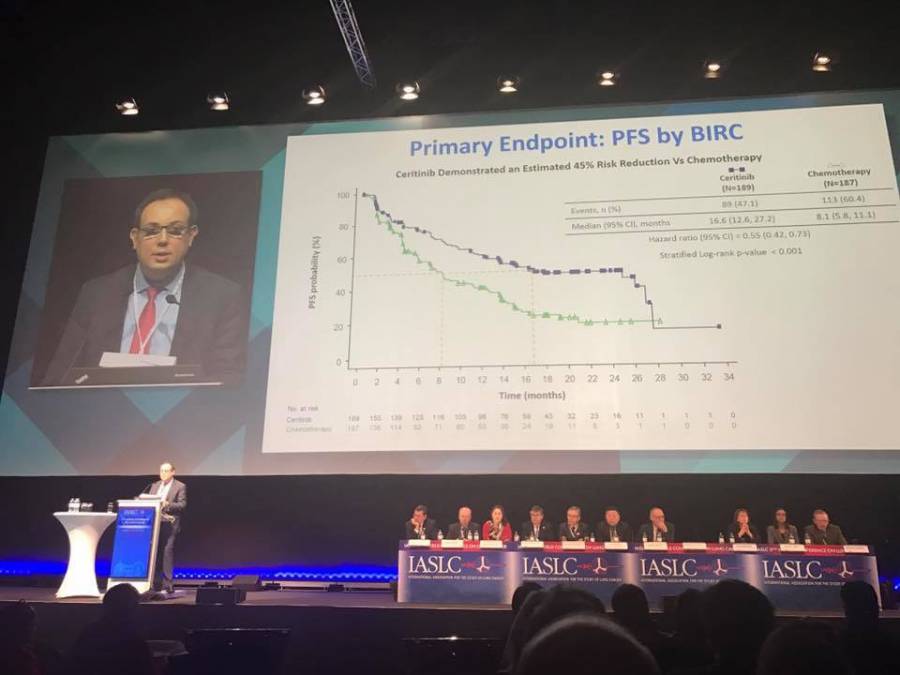
O deputado Hiran Gonçalves apresentou, na Comissão de Seguridade Social e Família da Câmara dos Deputados, substitutivo ao projeto de lei 7082/2017, que regulamenta a pesquisa clínica no Brasil. De acordo com o Dr. Fábio André Franke, vice-presidente da Sociedade Brasileira de Oncologia Clínica (SBOC) para Pesquisa Clínica e Estudos Corporativos, foi um grande avanço, especialmente se considerado este período de mudanças no governo. “Estamos bastante esperançosos de que o PL seja votado e finalizado nesta Comissão”, afirma o oncologista. “O objetivo é que possamos, no novo governo, submetê-lo à última Comissão e, ainda em 2019, contar com esse marco regulatório para liberar as pesquisas clínicas no Brasil num tempo mais ágil, tanto na aprovação regulatória quanto na aprovação ética.”
O Dr. Fábio Franke, que também presidente a Aliança Pesquisa Clínica Brasil, apoiadora do projeto de lei, considera que a aprovação será uma grande conquista para toda a comunidade científica brasileira. “A lei permitirá, principalmente em oncologia, que o Brasil efetivamente receba os estudos com novos desenhos, tanto em fase mais avançada como os de fase 1 e 2, e isso fará com que parte dos pacientes brasileiros, que são os principais interessados, tenha acesso aos tratamentos de ponta em primeira mão, nos mesmos moldes dos europeus, americanos e dos grandes centros especializados do mundo”, explica.
Substitutivo
O deputado Hiran Gonçalves destaca que “os princípios gerais, as diretrizes e regras internacionais aplicáveis aos estudos científicos que utilizam o ser humano em alguma fase da prospecção foram incorporadas à legislação interna por meio da Resolução nº 196, de 10 de outubro de 1996, do Conselho Nacional de Saúde – CNS, atualmente vigente”. Por isso, afirma ele, “a ideia de disciplinar o tema em uma lei ordinária é, com efeito, melhor do ponto de vista da segurança jurídica”, pois “as leis são a espécie mais apropriada para a delimitação dos direitos e deveres das pessoas”.
Em seu relatório, Hiran Gonçalves descreve pontos que têm mobilizado maior atenção durante a tramitação do projeto. Um deles é em relação às instâncias de controle: a nacional, representada pela Comissão Nacional de Ética em Pesquisa (Conep); e local, representada pelos Comitês de Ética em Pesquisa (CEP). “Tais instâncias foram reproduzidas no PL, mantendo-se a atribuição da análise ética da pesquisa pelos comitês. A Conep continua com a competência recursal e regulamentar do sistema, além de acompanhar, fiscalizar e apoiar os CEPs, como já faz atualmente”, descreve.
Outro ponto destacado pelo deputado diz respeito à vinculação da Conep, atualmente ligada ao Conselho Nacional de Saúde (CNS). Por isso, no substitutivo, ele retoma texto anterior do projeto, que vincula a Conep ao Ministério da Saúde, na Secretaria de Ciência, Tecnologia e Insumos Estratégicos (SCTIE). “Tal mudança tem sido rejeitada pelas entidades ligadas ao CNS. Todavia, essa alteração foi muito discutida no Senado, tendo sido demonstrado que a vinculação ao Ministério da Saúde foi defendida amplamente pelos setores representativos da comunidade científica, que consideram não adequada a submissão da Conep ao Conselho Nacional de Saúde, o qual não possuiria estrutura adequada e condizente para assumir funções de alta tecnicidade, além de ser apontado como um dos responsáveis pelos problemas, em especial a morosidade, do sistema de análise ética das pesquisas”, reforça Hiran.
O terceiro ponto destacado pelo relator, que tem provocado muitas discussões ao longo da tramitação, é o da continuidade do tratamento pós-pesquisa clínica. O substitutivo, no Art. 39, estabelece que “antes do início do ensaio clínico, patrocinador e pesquisador submeterão à CONEP um plano específico para cada ensaio clínico apresentando e justificando a necessidade ou não de fornecimento gratuito do medicamento experimental pós-ensaio clínico”. Em parágrafo único desse artigo, o relator propõe que “caso necessário patrocinador e pesquisador poderão submeter à CONEP o plano estabelecido (..) com prazo determinado diferenciado para o fornecimento gratuito pós-ensaio clínico do medicamento experimental, que deverá ser igualmente avaliado e aprovado pelo CEP de acordo com os critérios estabelecidos nesta Lei”.
No Art. 43, o substitutivo de Hiran Gonçalves estabelece que “o pesquisador e o patrocinador avaliarão o momento em que o fornecimento pós-ensaio clínico do medicamento experimental deverá ser interrompido, mediante o estabelecido pelo planejamento do ensaio clínico, conforme o Art. 39”. Além disso, estabelece oito condições para a interrupção do fornecimento, entre elas: “após, no máximo, dois anos da disponibilidade comercial do medicamento experimental no país; ou após cinco anos da disponibilidade comercial do medicamento experimental no país destinado a doenças raras e ultrarraras, conforme estabelecido pela Organização Mundial de Saúde; ou quando o medicamento experimental estiver disponível na rede pública de saúde”.